Risk-based Quality Management:A Transformative Approach for Clinical Trial Monitoring
Nov 05, 2021
Risk-based Quality Management:A Transformative Approach for Clinical Trial Monitoring
A Deep Look at Tigermed RBQM Platform
November, 2021
With the release of new policies, remote intelligent clinical trials and risk-based monitoring are turning out to be hot topics of the moment. What exactly is this quality management system, widely referred to as “RBQM”? What are the advantages of centralized monitoring compared to traditional on-site monitoring methods? And how can Tigermed RBQM platform benefit sponsors and CROs?
Key Points
Centralized monitoring not only complements to on-site monitoring, but also helps reduce the frequency of on-site monitoring and assists in the identification of potentially problematic data, thus a strategy of on-site and off-site monitoring can reduce overall operation cost also bring more data quality and efficiency.
RBQM professional strategy and system application can improve overall performance of clinical trial and reduce or limit potential risks.
An Innovative Shift in Clinical Trial Monitoring Models
Previously, it was common to increase the source data verification (SDV) ratio to improve the quality of clinical data. However, many overseas studies have shown that a high SDV ratio does not necessarily mean good data quality.
Since ICH E6R2 (Chapter 5.0) published the need for Risk-Based Monitoring (RBM) in 2016, the FDA has been improving the RBM strategy. Since 2020, China has also started to fully implement the ICH principles and issued a new version of the GCP (Article 31), both of which clearly propose a systematic, risk-based approach to monitoring clinical trials. Centralized monitoring approach uses different monitoring strategies for different projects, which can improve both operational efficiency and data quality.
Not only can centralized monitoring complement on-site monitoring, it can also help reduce the frequency of on-site monitoring and help identify potentially problematic data, thus help suggest priorities for on-site monitoring with considering different level of risks in sites. With mutual benefits from remote and on-site monitoring, it is recommended that a combined model of both types of monitoring will improve the quality and efficiency of clinical trials and enhance the protection of patient rights.
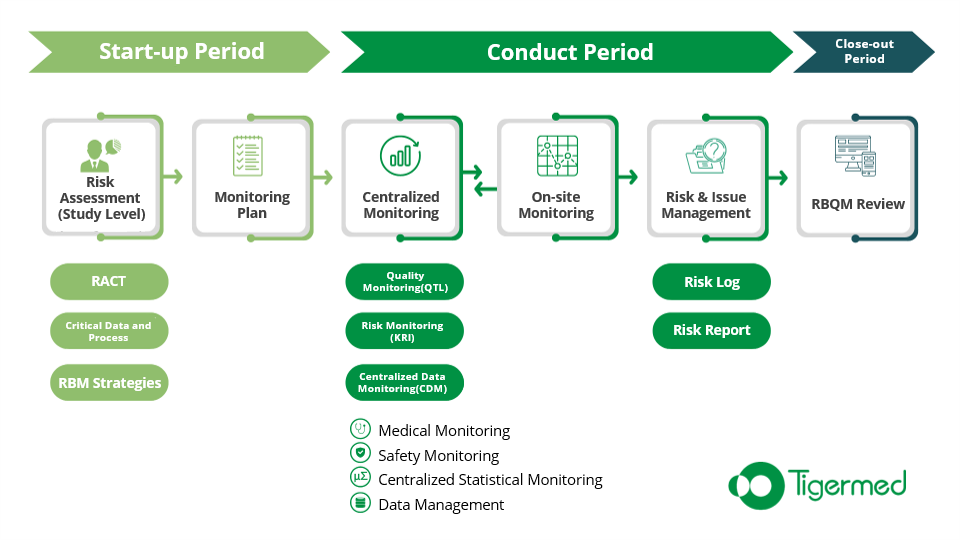
RACT: Evaluate Study Risks
As the first step in the implementation of RBQM, representatives of all functional departments (including operations, medicine, statistics, data management, drug safety, etc.) should be organized to conduct a full and comprehensive risk assessment of the study at the start-up stage, and guide the formulation of the quality management plan and strategy of the study. This evaluation step can be conducted with the Risk Assessment & Categorization Tool (RACT).
Target A: Define the scope of risks and responsibilities
During the risk assessment, the nature, source and potential causes of risks that may affect critical data collection or critical processes need to be identified and measures to control the risks need to be developed. All collaborators involved in the clinical trial need to communicate in a timely manner to ensure openness and transparency in risk management.
The assessment of risk should be considered at two levels.
System level, including facilities and equipment, SOPs, data system platforms, personnel, etc. For example, using good suppliers with well-trained project members in order to reduce the lower risk of the trials.
Study level, including trial drugs, trial design, data collection and recording, etc. For example, study patients take their own medications have a higher risk of problems in medication adherence than trials in which nurses administer injections, require more attention and control measures.
Target B: Focus on quality management
When study Protocols are developed, key aspects and data to protect the rights and safety of subjects and to ensure reliable clinical trial results should be clearly identified. Well-defined critical data and processes can guide quality management, including selecting key risk indicators, developing more targeted SDV strategies, and adjusting the focus of data management. For example, for supporting data (non-critical data), a more simplified approach can be used, which focused on logical checks, While for critical data, a combined approach, such as a combination of logical checks, statistical analysis and medical review, can be used to safeguard data integrity.
Target C: Assessing the overall risk level of the study
In addition to the qualitative description of the risk scope, the risk assessment also includes a quantitative assessment of the risk. The quantitative assessment should consider three aspects of risk, Probability, Detectability and Impact, and rate them on a number of degree scales (usually 3 – 5 scales), e.g. High: 3; Medium: 2; Low: 1.
Probability: The likelihood of errors occurring.
Detectability: The limit of errors that can be detected.
Impact: The impact of the error on the protection of the subject’s rights and on the reliability of the trial results.
The quantitative assessment results of all risk assessment questions were combined to derive the risk rating for each major risk category and the trial as a whole. Based on the estimated overall risk level, the sponsor can develop a more targeted monitoring strategy, including adjustments to the SDV /SDR (Source Data Verification / Source Data Review) work plan and the frequency of on-site monitoring.
KRI :Regular Monitoring of Risks
Key Risk Indicators (KRI) are Key Risk indicators that identify changes in a Risk area and can be monitored regularly. KRI is designed to effectively identify and monitor potential problems during clinical trial execution, guide quality focus, and intervene before risks become problems, so as to prevent problems from occurring or worsening, and ultimately protect patients’ rights and improve data quality.
QTL:Getting to the Heart of Clinical Data
The sponsor should preset the quality tolerance limits (QTL) and the corresponding remediation plan, and should summarize the deviations and remedial measures in the CSR.
The QTL should be set with reference to historical data from similar trials, and medical and statistical experts in the relevant fields should determine the QTL parameters for the trial and the definition of the parameters. The number of QTL parameters should not be excessive, and it is recommended to set 3-5 parameters that best reflect the safety of the trial subjects and data integrity.
QTL reflects the acceptable level of data variation for the parameters and using statistical methods can be used to define the level of abnormal variation.
CDM:Taking the High Ground
On-site monitoring is limited by cost and geographic location, and the number of monitoring is limited; whereas centralized monitoring is relatively weakly affected by cost and geographic location, and can theoretically increase the frequency of centralized data monitoring (CDM) indefinitely under the premise of using mature data systems, which can detect some risks more effectively in real time.
Typically, CDM includes medical assessment of subject enrollment, Protocol deviations, clinical endpoints, and medical review of safety data. Using the centralized data analysis platform, visualization of data with standard format (e.g., Protocol deviations and safety data) can help medical intuition quickly grasp the relationships inherent in the data and the areas of concern.
In addition, the application of statistical methods to monitor centralized data (Centralized Statistical Monitoring, CSM) can greatly enhance the comprehensiveness and penetration of data verification. Based on the fact that all centers participating in the same trial follow the same study Protocol, their clinical data should follow the same distribution. Through between sites and within sites statistical analysis of clinical data, the distribution of data anomalies can be examined to identify potential high-risk study sites, and then the integrity of the data can be analyzed in depth and in a targeted manner.
Commonly used methods for centralized statistical monitoring include analysis of dispersion of univariate data, the presence of digital preferences, and correlations among multivariate data. Correlation graphs include box plots, histograms, stem-and-leaf plots, Mahalanobis distances, and face plots.
Outlook
Clinical trials are a complex and rigorous project. In order to get more real and effective, safe new drugs to the people earlier, clinical researchers have invested a lot of effort to ensure that the trials are completed with high quality. At the same time, the explosive growth of the innovative drug industry has led to a rapid growth of pharmaceutical talent still in short supply.
As clinical trials continue to evolve towards patient decentralization and data center, clinical researchers continue to innovate and digital infrastructure continues to mature, data technology will play more and more important role in helping clinical researchers focus on higher value work and improve trial quality and efficiency, ultimately accelerating new drug development.
The site uses cookies in order to collect data to provide general statistics to optimize
site functionality and offer you a better experience. For more information, visit our Privacy Policy.